
БНБ
"БСЭ" (95279)
- Photogallery
- Естественные науки - Математика - Технология
- Авиация и машиностроение
- Высокие технологии
- Вычислительная техника
- Нанотехнология
- Роботехника
- Энергетика
- Электроника
Катализ
Определение "Катализ" в Большой Советской Энциклопедии
 |
| Катализ |
Воздействие катализатора открывает новый реакционный путь, обычно с большим числом стадий, на котором катализатор входит в состав активного комплекса (активированного комплекса) по крайней мере одной из стадий. Если при этом скорость реакции становится больше, чем в отсутствие катализатора, то Катализ называется положительным (его нередко отождествляют с общим понятием Катализ). Возможен и обратный случай, когда происходит отрицательный Катализ: в присутствии катализатора исключается один из возможных путей реакции и остаются лишь более медленные, в результате чего реакция замедляется или даже практически полностью подавляется (см. Антиокислители, Ингибиторы химические). Особый случай Катализ — ускорение реакции при воздействии продукта реакции или одного из промежуточных веществ, образующихся при реакции (см. Автокатализ).
Катализ не связан с изменением свободной энергии катализатора, и воздействие катализатора не может поэтому смещать положение равновесия химической реакции. Вблизи состояния равновесия катализаторы в равной степени ускоряют как прямую, так и обратную реакцию.
Основным фактором, определяющим скорость химического превращения, является энергия активации (Е) — разность энергий активного комплекса и исходных реагирующих молекул. Если предположить, что реакция не нарушает равновесного распределения энергии между молекулами, то вероятность образования активного комплекса, а следовательно, и скорость реакции в первом приближении пропорциональна exp (—E/RT), где R — газовая постоянная, Т — абсолютная температура. Отсюда следует, что скорость реакции тем больше, чем меньше Е, и вследствие экспоненциальной зависимости возрастает значительно даже при небольшом снижении Е. На рис. представлено изменение энергии при реакции без катализатора (кривая 1) и при участии катализатора (кривые 2 и 3). Кривая 2 с двумя максимумами соответствует образованию одного промежуточного продукта. Число стадий и промежуточных продуктов часто бывает значительно большим. Взаимодействие реагирующих веществ с катализатором может и не приводить к образованию стабильной формы промежуточного соединения (кривая 3). Но и в этом случае катализатор входит в состав активного комплекса и взаимодействие реагирующих веществ с катализатором определяет реакционный путь. Если энергии активных комплексов всех стадий реакционного пути с участием катализатора ниже энергии активного комплекса реакции без катализатора (т. е. 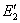 ,
, 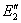 и E3 ниже E1), то участие катализатора приведёт к увеличению скорости (положительный Катализ). Во многих случаях Катализ ускорение реакции достигается благодаря появлению богатых энергией частиц в процессе самой реакции, причём их концентрация может превосходить равновесную (см. Цепные реакции). Например, каталитическое воздействие воды на окисление окиси углерода связано с развитием реакционных путей с участием гидроксильных групп и атомов водорода. Отрицательный Катализ часто связан с прекращением цепной реакции вследствие обрыва цепей при взаимодействии отрицательного катализатора с активными частицами. Примером может служить замедляющее влияние кислорода на соединение водорода с хлором.
и E3 ниже E1), то участие катализатора приведёт к увеличению скорости (положительный Катализ). Во многих случаях Катализ ускорение реакции достигается благодаря появлению богатых энергией частиц в процессе самой реакции, причём их концентрация может превосходить равновесную (см. Цепные реакции). Например, каталитическое воздействие воды на окисление окиси углерода связано с развитием реакционных путей с участием гидроксильных групп и атомов водорода. Отрицательный Катализ часто связан с прекращением цепной реакции вследствие обрыва цепей при взаимодействии отрицательного катализатора с активными частицами. Примером может служить замедляющее влияние кислорода на соединение водорода с хлором.
Характер промежуточного химического взаимодействия при Катализ весьма разнообразен. Обычно различают две группы каталитических процессов: кислотно-основной (гетеролитический) и окислительно-восстановительный (гомолитический). В процессах первой группы происходит промежуточное кислотно-основное взаимодействие реагирующих веществ с катализатором, например переход протона от катализатора к реагирующим веществам или наоборот. На последующих стадиях протон перемещается в обратном направлении, и катализатор восстанавливает свой состав. При Катализ апротонными кислотами взаимодействие осуществляется через свободную пару электронов реагирующего вещества. Примерами кислотно-основного Катализ могут служить гидролиз сложных эфиров, ускоряемый кислотами; гидратация олефинов в присутствии фосфорно-кислотных катализаторов; изомеризация и крекинг углеводородов на алюмосиликатных катализаторах; алкилирование; полимеризация и многие другие реакции. При реакциях окислительно-восстановительного Катализ промежуточное взаимодействие связано с электронными переходами между катализатором и реагирующими веществами. К этой группе относятся окисление двуокиси серы в трёхокись в производстве серной кислоты; окисление аммиака до окиси азота при получении азотной кислоты; многочисленные процессы парциального окисления органических соединений, например этилена в окись этилена, нафталина во фталевый ангидрид; гидрогенизация; дегидрогенизация; циклизация и ароматизация углеводородов; разложение перекиси водорода и многие др. Каталитической активностью в отношении окислительно-восстановительных реакций обладают преимущественно металлы 4-, 5- и 6-го периодов системы Д. И. Менделеева, имеющие недостроенную d-оболочку электронов, их соединения и в меньшей мере соединения элементов с достраивающейся f-оболочкой (лантаноиды и актиноиды).
Рассмотренные группы далеко не охватывают всё разнообразие каталитических реакций. Характер промежуточного взаимодействия при Катализ гораздо более сложен и зависит от всех деталей электронной структуры как реагирующих веществ, так и катализатора. Конкретные механизмы каталитических реакций многообразны и пока лишь в немногих случаях твёрдо установлены.
В зависимости от фазового состояния реагирующих веществ и катализатора различают гомогенный и гетерогенный Катализ Промежуточное положение занимает микрогетерогенный Катализ в коллоидных системах (например, Катализ ферментами). При гомогенном Катализ катализатор и реагирующие вещества образуют одну однородную систему, границы раздела между катализатором и реагирующими веществами отсутствуют. При гетерогенном Катализ катализатор и реагирующие вещества находятся в разных фазах и отделены друг от друга границей раздела. Наиболее важны случаи, когда катализатор является твёрдым телом, а реакционная система образует жидкую или газообразную фазу. Промежуточное взаимодействие происходит при этом преимущественно на поверхности твёрдого катализатора.
Выбор состава катализатора для определённой реакции является очень сложной проблемой, решаемой пока главным образом эмпирическим путём. В СССР предложен и развит ряд теоретических подходов, основанных на корреляции отдельных частных свойств катализаторов с их активностью. Так, мультиплетная теория Катализ (первые публикации 1929) предполагает промежуточное взаимодействие реагирующих веществ с несколькими атомами на поверхности твёрдых катализаторов и придаёт решающее значение соответствию расстояний между атомами в молекулах реактантов и параметров кристаллической структуры катализатора. В дальнейшем теория была дополнена представлением о необходимости определённого соответствия энергий связей, разрывающихся и образующихся в результате реакции, и энергий связей реактантов с катализатором при промежуточном взаимодействии. Значительное распространение в 50-х гг. получило представление о зависимости каталитической активности твёрдых катализаторов, обладающих полупроводниковыми свойствами, от их электрических характеристик, — так называемая электронная теория Катализ По этой теории предполагается, что промежуточное взаимодействие реактантов с катализатором осуществляется при участии электронов проводимости твёрдого катализатора и поэтому зависит от его коллективных электронных свойств — расположения энергетических зон и локальных уровней электронов, работы выхода электрона, концентрации носителей тока и др. В гетерогенном Катализ широко использовалось предположение (выдвинутое в 1939) о существовании на поверхности твёрдых катализаторов особых активных центров, представляющих собой ребра, углы или различные структурные нарушения (дислокации) нормальной кристаллической структуры. Предполагалось также, что при нанесении каталитически активного вещества на инертный носитель особые каталитические свойства проявляют отдельно расположенные атомы или совокупности небольшого числа атомов — ансамбли.
Появление точных методов определения поверхности катализаторов позволило установить, что активность, отнесённая к единице поверхности (удельная каталитическая активность), определяется химическим составом и очень мало зависит от структурных дислокаций. Удельная каталитическая активность различных граней кристаллов иногда различается в несколько раз. Большое влияние на активность оказывают нарушения химического состава (отклонение от стехиометрии, внедрение примесей, локальные химические образования и т.п.).
В 60-е годы промежуточное химическое взаимодействие в гетерогенном Катализ рассматривается преимущественно как локальное, определяемое электронной структурой отдельных атомов или ионов каталитически активного компонента на поверхности катализатора с учётом влияния ближайшего окружения. Значительную помощь в развитии этого подхода оказала обнаруженная экспериментально аналогия в действии твёрдых катализаторов, содержащих определённый металл, при гетерогенном Катализ и растворимых комплексов, компонентом которых является тот же металл, при гомогенном Катализ в растворах. При этом используются теории кристаллического поля и поля лигандов, ещё ранее успешно применявшиеся в химии комплексных соединений. Для ряда классов катализаторов и каталитических реакций установлены корреляции между каталитической активностью и энергиями связей реактантов с катализатором при промежуточном взаимодействии, облегчающие в отдельных случаях подбор катализаторов.
Первые научные сведения о Катализ относятся к началу 19 в. В 1806 французские химики Н. Клеман и Ш. Дезорм открыли каталитическое действие окислов азота на окисление сернистого газа в камерном процессе получения серной кислоты, В 1811 русский химик Катализ С. Кирхгоф открыл, что разбавленные кислоты способны вызывать превращение крахмала в сахар (глюкозу); в 1814 им же было установлено, что эту реакцию может катализировать диастаза из ячменного солода, — так было положено начало изучению биологических катализаторов — ферментов. В 1818 французский химик Л. Тенар установил, что большое число твёрдых тел оказывает ускоряющее действие на разложение растворов перекиси водорода, а английский химик Г. Дэви открыл способность паров спирта и эфира окисляться кислородом на платине. В 1822 нем. химик И. Дёберейнер установил, что водород и кислород соединяются на платине при обычной температуре. За этим последовало открытие и ряда др. примеров резкого положительного действия веществ на скорость или возникновение химических реакций. Это привело к выделению особой группы явлений, названных нем. химиком Э. Мичерлихом контактными (1833) и швед. химиком И. Берцелиусом каталитическими (1835).
В дальнейшем было открыто большое число каталитических реакций, и за последние 50 лет Катализ стал ведущим методом осуществления химических реакций в промышленности. Применение катализаторов позволяет проводить химические превращения с высокими скоростями при небольших температурах — большинство промышленных каталитических процессов без катализаторов вообще не могло бы быть реализовано. Подбирая катализаторы, можно направлять химические превращение в сторону образования определённого продукта из ряда возможных. Применение стереоспецифичных катализаторов позволяет регулировать и строение конечных продуктов, например полимеров. С помощью Катализ в начале 20 в. была решена проблема фиксации азота воздуха. Промотированные железные и другие катализаторы позволили преодолеть химическую инертность элементарного азота и осуществить синтез аммиака. Одновременно был разработан каталитический метод получения азотной кислоты путём окисления аммиака на платиновых сетках. На каталитических реакциях основываются современные методы получения водорода из природного газа. Каталитические методы занимают господствующее положение и в технологии нефтепереработки. Сотни миллионов тонн высококачественного моторного топлива производятся с помощью каталитических реакций крекинга, гидрокрекинга, риформинга, циклизации и изомеризации углеводородов нефти. Особенно большую роль играют каталитические методы в осуществлении процессов органического синтеза. В нашей стране впервые в мире было разработано и реализовано производство синтетического каучука, основанное на превращении этилового спирта в дивинил с помощью многокомпонентного окисного катализатора Лебедева. Каталитические методы используются для получения подавляющего большинства продуктов нефтехимического синтеза: растворителей, ароматических углеводородов, мономеров для производства синтетических каучуков, синтетических волокон и др. полимерных материалов. Катализаторы широко используются и для полимеризации.
Катализ играет ведущую роль в химических превращениях в живой природе. Вся сложная система управления жизненными процессами в организмах основана на каталитических реакциях. Биологические катализаторы, называемые ферментами или энзимами, представляют собой вещества белковой природы с химически активными группами, часто включающими в свой состав атомы переходных элементов. По некоторым свойствам ферменты превосходят промышленные катализаторы. В СССР и за рубежом широко ведутся исследования новых типов сложных синтетических катализаторов — комплексных соединений, органических полупроводников, полимеров, характеризующихся более простым составом по сравнению с ферментами, но моделирующих в известной степени их действие. Науке о Катализ принадлежит существенная роль как в прогрессе химической промышленности, так и в раскрытии важнейших биологических закономерностей.
Лит.: Баландин А. А., Мультиплетная теория катализа, ч, 1—2, М., 1963—64; Волькенштейн Ф. Ф., Электронная теория катализа на полупроводниках, М., 1960: Catalysis, ed. P. Н. Ernmett, v. 1—7, N. Y., 1954—60; Ашмор П.., Катализ и ингибирование химических реакций, пер. с англ., М., 1966; Томас Дж., Томас У., Гетерогенный катализ, пер. с англ.. М., 1969; Киперман С. Л., Введение в кинетику гетерогенных каталитических реакций, М., 1964; Боресков Г. Катализ, Катализ в производстве серной кислоты, М. — Л., 1954; Крылов О. В., Катализ неметаллами, Л., 1967; Основы предвидения каталитического действия. Труды IV Международного конгресса по катализу, т. 1—2, М., 1970.
Г. Катализ Боресков.

Изменение энергии реакционной системы вдоль пути реакции. А — исходное состояние; состояния, соответствующие образованию: В — промежуточного соединения, С — конечных продуктов, X1, X"2, Х"2, X3 — активных комплексов.
| "БСЭ" >> "К" >> "КА" >> "КАТ" >> "КАТА" |
Статья про "Катализ" в Большой Советской Энциклопедии была прочитана 1439 раз
| Бургер двойного помола |
| Сингапурский салат |
TOP 20
- Лемке Михаил Константинович
- Сульфгидрильные группы
- «Казарменный коммунизм»
- Японское море
- Периодическая система элементов
- Башкирская Автономная Советская Социалистическая Республика
- Объединённая партия гаитянских коммунистов
- Глициния
- Иммунитет (историч.)
- Андаманское море
- Сенсуализм
- Балкано-кавказская раса
- «Сообразительный»
- Звёздная астрономия
- Навигация (морск.)
- Ямполь (пос. гор. типа в Донецкой обл.)
- Мандельштама - Бриллюэна рассеяние
- Театральные учебные заведения
- Кульчицкая Елена Львовна
- Электрическая постоянная