
БНБ
"БСЭ" (95279)
- Photogallery
- Естественные науки - Математика - Технология
- Авиация и машиностроение
- Высокие технологии
- Вычислительная техника
- Нанотехнология
- Роботехника
- Энергетика
- Электроника
Органическая химия
Определение "Органическая химия" в Большой Советской Энциклопедии
Органическая химия, раздел химии, естественнонаучная дисциплина, предметом изучения которой являются соединения углерода с др. элементами, называемые органическими соединениями, а также законы превращения этих веществ. Углерод образует соединения с большинством элементов и обладает наиболее выраженной способностью по сравнению с др. элементами к образованию молекул цепного и циклического строения. Скелет таких молекул может состоять из практически неограниченного числа атомов углерода, непосредственно соединённых друг с другом, или включать наряду с углеродом атомы др. элементов. Для соединений углерода наиболее характерно явление изомерии, т. е. существование веществ, одинаковых по составу и молярной массе, но различающихся последовательностью сцепления атомов или расположением их в пространстве и вследствие этого по химическими и физическим свойствам. В результате этих особенностей число органических веществ чрезвычайно велико, к 70-м гг. 20 в. известно более 3 млн., в то время как соединений всех остальных элементов - немногим более 100 тыс.
Органические соединения способны к сложным и многообразным превращениям, существенно отличным от превращений неорганических веществ, и играют основную роль в построении и жизнедеятельности растительных и животных организмов. К органическим соединениям относятся углеводы и белки, с которыми связан обмен веществ; гормоны, регулирующие этот обмен; нуклеиновые кислоты, являющиеся материальными носителями наследственных признаков организма; витамины и др. Органическая химия представляет собой т. о. как бы своеобразный «мост» между науками, изучающими неживую материю и высшую форму существования материи - жизнь. Многие явления и закономерности химической науки, например изомерия, впервые были открыты при изучении именно органических соединений.
Классификация органических соединений. Все органические соединения подразделяются на три основных ряда, или класса: ациклические, изоциклические и гетероциклические. К первому классу (жирных, или алифатических) соединений относят углеводороды и их производные с незамкнутыми цепями: гомологический ряд метановых углеводородов, называемый также рядом насыщенных углеводородов, или алканов; гомологические ряды ненасыщенных углеводородов - этилена (алкенов), ацетилена (алкинов), диенов и др. (см. Ациклические соединения). К классу изоциклических (карбоциклических) соединений относят углеводороды и их производные, в молекулах которых имеются циклы из атомов углерода: углеводороды и их производные циклопарафинового, или полиметиленового, ряда, циклические ненасыщенные соединения (см. Алициклические соединения, Циклоалканы), а также ароматические углеводороды и их производные, содержащие бензольные ядра (в частности, и многоядерные ароматические соединения, например, нафталин, антрацен). К классу гетероциклических соединений относят органические вещества, в молекулах которых имеются циклы, содержащие, кроме углерода, атомы О, N, S, Р, As или др. элементов.
От каждого углеводорода образован отдельный генетический ряд (см. Гомологические ряды), представители которого формально производятся путём замены атома водорода в углеводороде той или иной функциональной группой, определяющей химические свойства соединения. Так, в генетический ряд метана CH4 входят хлористый метил CH3Cl, метиловый спирт CH3OH, метиламин CH3NH2, нитрометан CH3NO2 и др. Аналогично представители генетического ряда бензола C6H6 - хлорбензол C6H5Cl, фенол C6H5OH, анилин C6H5NH2, нитробензол C6H5NO2 и др. Одноимённо замещённые представители различных генетических рядов составляют гомологические ряды производных: галогенсодержащих соединений, спиртов, аминов, нитросоединений и др.
О названиях органических соединений см. Номенклатура химическая.
Историческая справка. Истоки Органическая химия восходят к глубокой древности (уже тогда знали о спиртовом и уксуснокислом брожении, крашении индиго и ализарином). Однако ещё в средние века (период алхимии) были известны лишь немногие индивидуальные органические вещества. Все исследования этого периода сводились главным образом к операциям, при помощи которых, как тогда думали, одни простые вещества можно превращать в другие. Начиная с 16 в. (период ятрохимии) исследования были направлены в основном на выделение и использование различных лекарственных веществ: был выделен из растений ряд эфирных масел, приготовлен простой диэтиловый эфир, сухой перегонкой древесины получены древесный (метиловый) спирт и уксусная кислота, из винного камня - винная кислота, перегонкой свинцового сахара - уксусная кислота, перегонкой янтаря - янтарная. Большая роль в становлении Органическая химия принадлежит А. Лавуазье, который разработал основные количественные методы определения состава химических соединений, в дальнейшем последовательно улучшенные Л. Тенаром, Й. Берцелиусом, Ю. Либихом, Ж. Дюма. Принципы этих методов (сожжение навески вещества в атмосфере кислорода, улавливание и взвешивание продуктов сгорания - CO2 и H2O) лежат в основе современного элементного анализа, в том числе и микроанализа. В результате анализа большого числа различных веществ доминировавшее ранее представление о принципиальном различии веществ растительного и животного происхождения постепенно отпадало.
Впервые название «органические соединения» встречается к конце 18 в. Термин «Органическая химия» был введён Берцелиусом в 1827 (в написанном им первом руководстве по Органическая химия). Явление изомерии было открыто Ф. Вёлером и Либихом в 1822-23. Первый синтез органического вещества осуществил Вёлер, получивший в 1824 щавелевую кислоту из дициана и в 1828 - мочевину нагреванием циановокислого аммония. Начиная с середины 19 в. число органических веществ, получаемых синтетически, быстро возрастает. Так, в 1842 Н. Н. Зинин восстановлением нитробензола получил анилин, в 1845 А. Кольбе синтезировал уксусную кислоту, в 1854 П. Бертло - вещества типа жиров. В 1861 А. М. Бутлеров получил первое искусственное сахаристое вещество, названное им метиленитаном, из которого впоследствии была выделена акроза. Синтетическое направление в Органическая химия приобретает всё большее значение. В результате успехов синтеза господствовавшее идеалистическое представление о необходимости «жизненной силы» для создания органических веществ (см. Витализм) было отвергнуто.
Теоретические представления в Органическая химия начали развиваться со 2-й четверти 19 в., когда была создана радикалов теория (Либих, Вёлер, Э. Франкленд, Р. Бунзен и др.). Основное её положение о переходе группы атомов - радикалов из одного соединения в другое в неизменном виде остаётся в большом числе случаев справедливым и в настоящее время. На этом представлении основаны многие физические и химические методы исследования веществ неизвестной структуры. Впоследствии (1834-39) Дюма показал возможность замещения положительно заряженных атомов в радикале на электроотрицательные без серьёзных изменений электрохимического характера радикала, что до Дюма считалось невозможным.
На смену теории радикалов пришла типов теория (1848-51, 1853), созданная Дюма, Ш. Жераром и О. Лораном. Последним удалось классифицировать органические вещества по типам простейших неорганических соединений. Так, спирты считались соединениями типа воды, амины - типа аммиака, галогеналкилы - типа хлористого водорода. Позднее Ф. А. Кекуле установил четвёртый тип - тип метана, от которого он производил все углеводороды. Теория типов позволила создать чёткую классификацию органических соединений, которая лежит в основе современной классификации органических веществ. Однако эта теория стремилась лишь к объяснению реакционной способности органических веществ и отрицала принципиальную возможность познания их строения. В 1853 Франкленд, изучая металлоорганические соединения, ввёл представление о валентности. В 1857 Кекуле высказывает мысль о возможности сцепления атомов углерода друг с другом и доказывает четырёхвалентность углерода. В 1858 А. Купер, используя правило валентности и положение Кекуле о сцеплении атомов углерода, впервые отходит от теории типов и пишет формулы органических веществ, очень близкие к современным. Однако идеи теории типов оставались ещё очень сильны и создание теории продолжало отставать от развития эксперимента.
В 1861 Бутлеров создал химического строения теорию органических веществ. Он ввёл в Органическая химия ряд новых понятий: о химической связи, порядке связей атомов в молекуле, о взаимном влиянии атомов, непосредственно связанных или не связанных друг с другом, и др. Теория строения Бутлерова блестяще объяснила остававшиеся непонятными известные к тому времени случаи изомерии. В 1864 Бутлеров предсказал возможность изомерии углеводородов и вскоре (1867) подтвердил это синтезом изобутана. Созданное Бутлеровым стройное учение лежит в основе современных представлений о химической строении органических веществ. Одно из важнейших положений теории строения - о взаимном влиянии атомов - впоследствии было развито В. В. Марковниковым. Детальное изучение этого влияния способствовало дальнейшему развитию теории строения и представлений о распределении электронной плотности и о реакционной способности органических соединений.
В 1869 И. Вислиценус показал, что явление изомерии наблюдается и при совершенно одинаковой последовательности сцепления атомов в молекуле. Он доказал идентичность строения обычной молочной кислоты и мясо-молочной и пришёл к выводу, что тонкие различия в свойствах молекул с одинаковой структурой следует искать в различном расположении их атомов в пространстве. В 1874 Я. Вант-Гофф и французский химик Ж. Ле Бель создали теорию пространств. расположения атомов в молекуле - стереохимию. В основе этой теории, по Вант-Гоффу, лежит представление о тетраэдрической модели четырёхвалентного атома углерода и о том, что оптическая изомерия является следствием пространственной асимметрии молекулы, в которой атом углерода соединён с четырьмя различными заместителями (см. Асимметрический атом). Вант-Гофф высказал также предположение о возможности др. вида пространственной изомерии при отсутствии в молекуле асимметричного атома углерода. Вскоре Вислиценус доказал, что фумаровая кислота, которую ранее считали полимером малеиновой кислоты, представляет собой её геометрический изомер (геометрическая, или цис-транс-изомерия). Ясно, что стереохимическое учение могло быть создано только на основе представлений о строении (структуре) молекулы в бутлеровском понимании.
К конце 19 в. накопился большой фактический материал, в том числе и по ароматическим соединениям; в частности, широко была изучена химия бензола, открытого М. Фарадеем в 1825. Первая т. н. «бензольная теория» строения ароматических соединений была создана в 1865 Кекуле. В ней высказывается мысль о том, что атомы углерода в органических соединениях могут образовывать кольца. Согласно этой теории, бензол обладает симметричной структурой вследствие кольцеобразного строения сцепленных попеременно простыми и двойными связями шести метиновых СН-групп. Однако, исходя из строения бензола по Кекуле, следовало допустить наличие двух орто-замещённых гомологов или производных бензола, чего на самом деле не наблюдалось. Устойчивость бензола к сильным окислителям и некоторые др. т. н. ароматическим свойства бензола и его производных также противоречили предложенной формуле. Поэтому Кекуле ввёл (1872) представление об осцилляции (быстром перемещении) двойных связей и устранил формальные различия между двумя орто-положениями. Несмотря на то, что строение бензола по Кекуле находилось в противоречии с данными о его физических и химических свойствах, оно долгое время без всяких изменений принималось подавляющим числом химиков. Т. о., остался ряд вопросов, не разрешимых с точки зрения «классической» теории строения. К этим вопросам относится и своеобразие свойств многих др. соединений с сопряжёнными системами связей. Строение бензола и др. ароматических систем могло быть установлено лишь с появлением физических методов исследования и с развитием квантово-химических представлений о строении органических веществ.
Электронные представления [В. Коссель (1916) и Г. Льюис (1916)] придали физическое содержание понятию химической связи (пара обобщённых электронов); однако в том виде, в каком они были сформулированы, эти представления не смогли отразить тонких переходов от ковалентной к ионной связи и в Органическая химия оставались в значительной степени формальными. Только с помощью квантово-химического учения было вложено принципиально новое содержание в правильные в основном представления электронной теории.
Представления Льюиса о паре электронов, образующих связь и всегда строго локализованных на этой связи, оказались приближёнными и в большинстве случаев не могли быть приняты.
Современные представления теории строения и значение Органическая химия Учёт квантовых свойств электрона, представления об электронной плотности и о взаимодействии электронов в сопряжённых системах открыли новые возможности для рассмотрения вопросов о строении, взаимном влиянии атомов в молекуле и о реакционной способности органических соединений (см. Электронные теории в органической химии, Квантовая химия). В насыщенных углеводородах одинарные связи С-С (s-связи) действительно реализуются парой электронов; в симметричных углеводородах электронная плотность в пространстве между соединившимися атомами С-С больше суммы соответствующих электронных плотностей тех же изолированных атомов и симметрично распределена относительно оси, соединяющей центры атомов. Увеличение электронной плотности - результат перекрывания электронных облаков атомов по прямой, соединяющей их центры. В несимметричных парафинах появляется возможность неполной симметрии в распределении электронной плотности; однако эта асимметрия столь незначительна, что дипольные моменты всех парафиновых углеводородов почти не обнаруживаются. То же касается и симметрично построенных непредельных углеводородов (например, этилена, бутадиена), у которых атомы С соединены друг с другом двойной связью (s- и p-связью). Введение в молекулы этих веществ электронодонорной метильной группы вследствие высокой поляризуемости p-связи приводит к смещению электронной плотности к крайнему атому углерода, и пропилен (I) уже имеет дипольный момент 0,35 Д, а 1-метилбутадиен - 0,68 Д. Распределение электронной плотности в этих случаях принято изображать одной из следующих схем: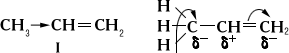
(Знаки d+ и d- показывают возникающие частичные заряды на атомах С)
В представления о распределении электронной плотности хорошо укладывается ряд эмпирических правил Органическая химия Так, из приведённой выше формулы пропилена следует, что при гетеролитическом присоединении к нему галогеноводородов протон должен фиксироваться в месте наибольшей электронной плотности, т. е. у наиболее «гидрогенизированного» атома углерода (см. Марковникова правило). Значительно сильнее сказывается введение в молекулы углеводородов атомов или групп, сильно отличающихся по электроотрицательности от атомов углерода или водорода. Например, введение электрофильного заместителя в молекулы углеводородов ведёт к изменению подвижности атомов водорода в связях С-Н, О-Н и др. Подобного рода взаимное влияние атомов, также объяснимое изменением распределения электронной плотности, быстро затухает у насыщенных соединений и почти без затухания передаётся по цепи сопряжённых связей (см. Сопряжение связей).
Принято различать два вида электронного влияния заместителей: индуктивное - по цепи s-связей и влияние сопряжения - по цепи с сопряжёнными связями. Так, увеличение кислотности хлоруксусной кислоты (II) по сравнению с уксусной СН3СООН объясняется индуктивным влиянием атомов хлора, а подвижность атомов водорода метильных групп в уксусном (III) или сорбиновом (IV) альдегиде - влиянием сопряжения: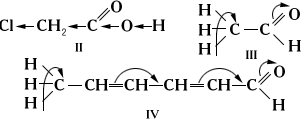
Перераспределение электронной плотности, особенно в момент реакции, происходит не только в связях, которые затрагиваются реакцией, но и в др. частях молекулы. Кажущаяся ненормальность солеобразования n-диметиламиноазобензола (V) с фиксацией протона слабоосновным атомом азота азогруппы объясняется перемещением реакционного центра молекулы вследствие сдвига электронной плотности в момент реакции в направлении, указанном стрелками: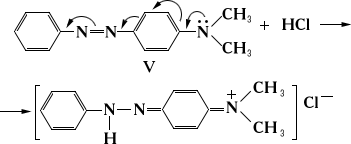
Влияние сопряжения проявляется и в тех случаях, когда два возможных направления реакции органических веществ не обусловлены таутомерией. Так, алкилирование натрийенолятов по атому углерода происходит вследствие перемещения реакционного центра благодаря сопряжению связей: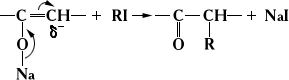
Взаимное влияние атомов в результате сопряжения связей проявляется также в ароматических соединениях (см. Ориентации правила). При электрофильном замещении электронодонорные (нуклеофильные) заместители (VI) ориентируют в орто- и пара-положения, электроноакцепторные (электрофильные) заместители (VII) - в мета-положение: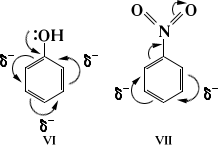
Т. о., на основании современных квантово-химических представлений разнообразные процессы Органическая химия нашли естественное объяснение. Теоретические представления Органическая химия окрепли и получили предсказательные возможности.
В результате развития теоретических и физических методов исследования был окончательно решен вопрос о строении ароматических систем, в том числе и бензола. Строение последнего описывается следующим образом: шесть атомов углерода бензольного кольца находятся в одной плоскости и соединены s-связями; шесть p-электронов составляют единую подвижную электронную систему. Следствием этого является полная подтверждаемая опытом равноценность связей С-С и высокая симметрия бензола с осью шестого порядка. Из этих положений следует, что бензол неполярен и обладает анизотропией диамагнитной восприимчивости. Аналогичными свойствами характеризуются все ароматические системы, у которых число p-электронов равно 4n + 2 (правило Хюккеля). Бензол - далеко не единичный пример соединений с выравненными двойными и простыми связями; аналогичная картина наблюдается у трополона, тропилийбромида, ферроцена, дифенилполиенов и др. Вполне удачного графического изображения строения бензола и др. ароматических соединений выработать не удалось. Для описания их строения используют набор валентных схем (VIII), впервые предложенных Л. Полингом в его резонанса теории, или систему обозначения (IX), где изогнутые стрелки показывают также выравненность связей (впервые применена в теории мезомерии):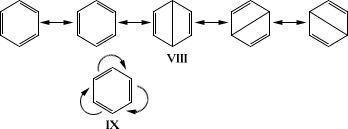
Этими же обозначениями пользуются для графической интерпретации равномерного распределения электронной плотности в симметричных ионах, например в карбоксилат-анионе (соответственно Х и XI), при объяснении слабоосновных свойств амидов кислот (XII и XIII) и в др. случаях: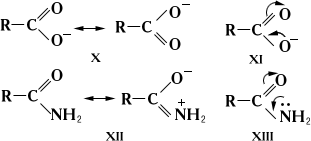
Приблизительно со 2-й половины 20 в. Органическая химия вступила в новую фазу. Многие направления её развивались столь интенсивно, что выросли в большие специализированные разделы, называется по научному или прикладному признаку (стереохимия, химия полимеров, природных веществ, антибиотиков, витаминов, гормонов, металлоорганических соединений, фторорганических соединений, красителей и др.).
Успехи теории и развитие физических методов исследования (например, рентгенографии молекул, ультрафиолетовой и инфракрасной спектроскопии, раманспектроскопии, ядерного магнитного резонанса, химически индуцированной динамической поляризации ядер, масс-спектрометрии), а также методов идентификации и разделения различных веществ с помощью хроматографии сделали возможным быстрый структурный анализ сложнейших органических соединений и быстрое решение многих важных проблем. Применение физических методов для исследования кинетики реакций органических веществ (см. Кинетика химическая) позволяет изучать реакции с периодом полупревращения 10-8-10-9 сек. Корреляционные уравнения, основанные на принципе линейности свободной энергии, дают возможность количественной оценки зависимостей между строением и реакционной способностью органических соединений, даже тех, которые обладают физиологическим действием.
Органическая химия оказалась тесно связанной со смежными естественными науками - биохимией, медициной и биологией, применение идей и методов Органическая химия в этих науках в значительной степени обусловило развитие нового направления - молекулярной биологии.
Методы Органическая химия наряду с физическими методами исследования сыграли важную роль в установлении строения нуклеиновых кислот, многих белков, сложных природных соединений; с их же помощью были раскрыты механизм и регуляция синтеза белков (см. Генетический код). Чрезвычайно возросли синтетические возможности Органическая химия, которые привели к получению таких сложно построенных природных веществ, как хлорофилл, витамин B12 (Р. Вудворт), полинуклеотиды с определённым чередованием звеньев (А. Тодд, Х. Г. Корана) и др. Огромный успех этих методов - разработка автоматического синтеза многих полипептидов, в том числе и ферментов.
Синтезирован новый класс органических соединений, образованных сплетением двух или более циклических молекул подобно обычной цепи (катенаны, на схеме слева) или подобно гантели, на ось которой надето кольцо (ротаксаны, справа):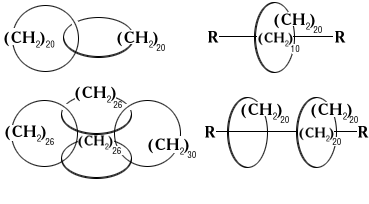
Отдельные части этих молекул связаны механическими силами. Наиболее значительным достижением синтетической Органическая химия и биохимии можно считать синтез гена, который осуществил Х. Г. Корана с сотрудниками (1967-70).
Большое значение приобрели методы Органическая химия в современной технологии производства каучуков синтетических, пластических масс, волокон синтетических, красителей, медикаментов, в промышленности кинофотоматериалов, стимуляторов роста растений, средств борьбы с вредителями сельского хозяйства (пестицидов) и многое др. Успехи Органическая химия в области основного органического синтеза и нефтехимического синтеза не только изменили технологию ряда производств, но и привели к созданию новых видов продукции. Благодаря установлению зависимостей между строением и свойствами органических соединений становится возможным создание новых материалов различных назначений с заранее заданными свойствами. Органическая химия достигла такого уровня, который отвечает её важной роли в создании материальной культуры современного общества.
Научные учреждения и организации, периодические издания. Научную работу по Органическая химия в СССР проводят научно-исследовательские институты АН СССР - институт органической химии им. Н. Д. Зелинского (ИОХ), институт органической и физической химии им. А. Е. Арбузова (ИОФХ), институт нефтехимической синтеза им. А. В. Топчиева (ИНХС), институт элементоорганич. соединений (ИНЭОС), институт химии природных соединений им. М. М. Шемякина (ИХПС); Сибирского отделения АН СССР - Новосибирский институт органической химии (НИОХ), Иркутский институт органический химии (ИИОХ), институт химии нефти; республиканскиъ академий - институты органической химии Армянской ССР, Киргизской ССР, УССР, институт тонкой органической химии им. А. Л. Мнджояна (Армянская ССР), институт физико-органической химии (БССР), институт физической и органической химии им. П. Г. Меликишвили (Грузинская ССР), институт органического синтеза (Латвийская ССР) и др.
Национальный комитет советских химиков является членом Международного союза чистой и прикладной химии (International Union of Pure and Applied Chemie, IUPAC, ЮПАК), который организует 1 раз в 2 года конгрессы, конференции и симпозиумы, в частности и по Органическая химия
Периодические издания, в которых публикуются работы по Органическая химия: в СССР - «Журнал органической химии» (с 1965); «Журнал общей химии» (с 1931); «Химия гетероциклических соединений» (Рига, с 1965); «Химия природных соединений» (Ташкент, с 1965); «Экспресс-информация. Промышленный органический синтез» (с 1960); за рубежом - «Journal of Organic Chemistry» (Wash., с 1936); «Journal of the Chemical Society. Perkin Transaction. 1. Organic and Bio-organic Chemistry» (L., с 1972); II. «Physical Organic Chemistry» (L., с 1972); «Justus Liebigs Annalen der Chemie» (Weinheim, с 1832); «Bulletin de la Societe chimique de France», pt. 2 (P., с 1858); «Journal of the Society of Organic Synthetic Chemistry of Japan» (Tokyo, с 1943); международные - «Tetrahedron» (N. Y., с 1957); «Tetrahedron Letters» (L., с 1959); «Synthesis» (Stuttgart, с 1969); «Synthetic Communication» (N. Y., с 1971); «Journal of the Organo-metallic Chemistry» (Lausanne, с 1964); «Journal of Heterocyclic Chemistry» (L., с 1964); «Organic Magnetic Resonance» (L., 1969); «Organic Mass Spectrometry» (L., 1968); «Organic Preparations and Procedures» (N. Y., с 1969). литература по Органическая химия реферируется в журналах: «Chemical Abstracts» (Easton, с 1907), реферативный журнал «Химия» (с 1953), «Chemisches Zentralblatt» (совместно ГДР и ФРГ, В., с 1830).
Лит.: Бутлеров А. М., Введение к полному изучению органической химии, в. 1-3, Каз.,1864-66; его же, Избр. работы по органической химии, М., 1951; Марковников В. В., Избр. труды, М., 1955; Гьельт Э., История органической химии с древнейших времён до настоящего времени, пер. с нем., Хар. - К., 1937; Шорлеммер К., Возникновение и развитие органической химии, пер. с англ., М., 1937; Джуа М., История химии, пер. с итал., М., 1966; Rodd’s chemistry of carbon compounds, 2 ed., v. 1-2, Amst. - [a. o.], 1964-1968; Beilsteins Handbuch der organischen Chemie, 4 Aufl., bearb. von B. Prager [u. a.], Bd 1-34, В., 1918-1944 (с 1928 года изд. доп. тт.); Houben-Weyl, Methoden der organischen Chemie, 4 Aufl., Bd 1-12, Stuttg., 1953-68; Краткая химическая энциклопедия, т. 1-5, М., 1961-67; Несмеянов А. Н., Несмеянов Н. А., Начала органической химии, т. 1-2, М., 1969-70; Неницеску К. Д., Органическая химия, пер. с рум., т. 1-2, М., 1962-63; Робертс Дж., Касерио М., Основы органической химии, пер. с англ., ч. 1-2, М., 1968; Физер Л., Физер М., Органическая химия, пер. с англ., М., 1966; Чичибабин А. Е.., Основные начала органической химии, т. 1-2, М., 1957-63; Ингольд К., Теоретические основы органической химии, пер. с англ., М., 1973; Перспективы развития органической химии, под ред. А. Тодда, пер. с англ., М., 1959.
И. Л. Кнунянц.
| "БСЭ" >> "О" >> "ОР" >> "ОРГ" |
Статья про "Органическая химия" в Большой Советской Энциклопедии была прочитана 1210 раз
| Коптим скумбрию в коробке |
| Кишки на гриле |
TOP 20
- Лемке Михаил Константинович
- Сульфгидрильные группы
- «Казарменный коммунизм»
- Японское море
- Периодическая система элементов
- Башкирская Автономная Советская Социалистическая Республика
- Объединённая партия гаитянских коммунистов
- Глициния
- Иммунитет (историч.)
- Андаманское море
- Сенсуализм
- Балкано-кавказская раса
- «Сообразительный»
- Звёздная астрономия
- Навигация (морск.)
- Ямполь (пос. гор. типа в Донецкой обл.)
- Мандельштама - Бриллюэна рассеяние
- Театральные учебные заведения
- Кульчицкая Елена Львовна
- Электрическая постоянная